PanGenie is a pangenome-based genotyper using short-read data. It computes genotypes for variants represented as bubbles in a pangenome graph by taking information of already known haplotypes (represented as paths through the graph) into account. It can only genotype diploid individuals. The required input files are described in detail below. 总结来说, 作者给出了两种方式来安装, Short Notes: pangenie需要的variants file有以下几个特点(其本质上是从构建pan-genome graph过程中产生的VCF中获得), multi-sample:包含多条resolvde haplotype,且至少有一条haplotype的sample info是已知的 fully-phased:举个例子,hifiasm过程中产生的两条phased haplotype即可 non-overlapping variants:由于基于pan-genome graph产生得到的variants在physical position上存在重叠,而这一类是不可以作为pangenie的输入数据的 sequence-resolved:需要清晰地保存了REF allele和ALT allele信息,像2015年Evan Eichler它们发布的结果不可以作为pangenie的输入文件(no information about REF & ALT) 针对第三点,pangenie官方给出的示意格式, 准备上述variants file的格式如下, also see Wiki for different ways to generate VCFs 1)vcfbub 2)针对mini-cactus分析得到的vcf,github.com/eblerjana/gen... 该pipeline的两种特点, 3)再者使用的是其他软件(e.g., PAV, developed by Evan),则可以使用pangenie自带的pipeline来进行merge和过滤, github.com/eblerjana/pan...,该流程基于snakemake Short Notes: 哪些VCF是无法满足上述数据的数据要求? Note again that the haplotypes must be phased into a single phased block. So phased VCFs generated by phasing tools like WhatsHap are not suitable! 结果文件, 1)additional genotype predictions; 2)genotype likelihoods; 3)genotype qualities;Intro|Pan-Genie能做什么?
Pangenie|软件安装
git clone https://github.com/eblerjana/pangenie.git
cd pangenie
conda env create -f environment.yml
conda activate pangenie
mkdir build; cd build; cmake .. ; make
# mamba activate pangenie
Pangenie|输入文件准备
01)pan-genome graph in variants format

# 如何过滤准备得到对应的pan-genie input vcf
vcfbub -l 0 -r 100000 --input <your-vcf-file> > pangenie-ready.vcf
02)short reads input
03)reference genome
Pangenie|demo
PanGenie-index -r test-reference.fa -v test-variants.vcf -o preprocessing -e 100000
PanGenie -f preprocessing -i test-reads.fa -o test -e 100000
test_genotyping.vcf:与输入variants保留着相同的REF & ALT,但是基于k-mer的genotyping算法重新估计了如下的三个参数,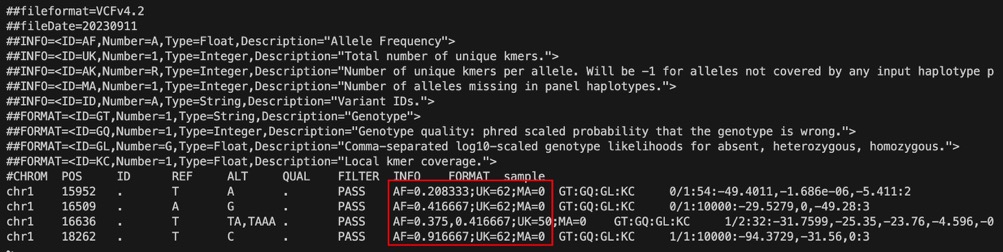
Hi,这里是有朴的第二大脑。
很高兴与你相遇
很高兴与你相遇